

全球最权威的蛋白质定向进化榜单
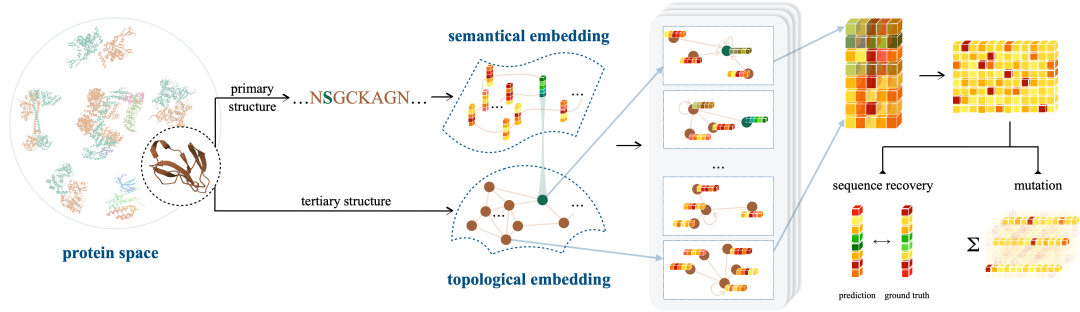
Tan Y, Zhou B, Zheng L, et al. Semantical and Topological Protein Encoding Toward Enhanced Bioactivity and Thermostability[J]. bioRxiv, 2023: 2023.12. 01.569522.

全球最权威的蛋白质定向进化榜单
Tan Y, Zhou B, Zheng L, et al. Semantical and Topological Protein Encoding Toward Enhanced Bioactivity and Thermostability[J]. bioRxiv, 2023: 2023.12. 01.569522.

